



华纳生物,DMF时代大潮砥砺奋进,为生物制品申报提速!
发布日期:2024-03-15 浏览次数:4705
2022年10月31日,国家药品监督管理局食品药品审核查验中心颁布了《细胞治疗产品生产质量管理指南(试行)》。该指南特别强调了治疗性起始物料的GMP资质问题,明确指出直接用于细胞产品生产的基因修饰载体或其他功能性材料(例如病毒、质粒、RNA等)的生产、检验和放行流程必须符合GMP及其相关附录的严格标准。
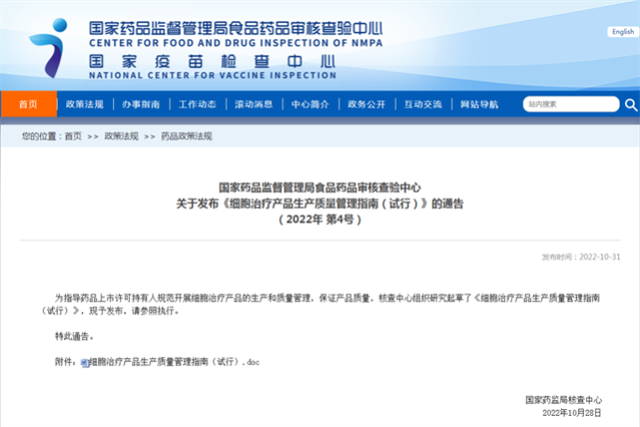
然而,国内当前的研发和注册环境中,研究者在选择病毒、质粒、RNA等关键物料的GMP级别时面临诸多限制。由于许多研究者对GMP级别物料的重要性认识不足,他们在注册过程中可能会随意选择关键物料用于临床药品生产。
我国生物药物的研究者向CDE(国家药品监督管理局药品审评中心)提交生物制品药物申报,如比如临床研究申请(IND)、新药注册(NDA)和生物制品许可证申请(BLA)等,必须遵循CTD(通用技术文档)格式来整理注册文件。这些文件需要提供药物在安全性、有效性和质量三大方面的详尽信息,涵盖原液生产工艺、制剂生产工艺以及关键活性物料的相关技术细节。然而,准备这些材料不仅费时费力,而且由于生物制品原液生产工艺的复杂性,涉及众多上下游工艺中的关键物料,这些物料的GMP资质问题往往成为申报过程中的一大难题。
缺乏关键物料的GMP认证资质渠道会严重拖慢临床申报的进度,为应对这一挑战,美国FDA(食品药品监督管理局)建立了生物制品关键物料的DMF(药品主文件)备案体系。通过这一体系,生物制品的研究者可以将原液生产所需的关键物料技术信息,以DMF文件的形式直接提交给FDA进行备案并获得备案编号。研究者在生物药研究项目中需要提交FDA临床申请或上市注册的监管备案文件时可直接引用相关DMF编号,从而节约产品审查和评估时间,缩减新药临床申报的准备工作,这有力促进了研究者的药物上市申报进程。
从DMF备案登记的范围来看,美国的DMF适用范围相对较广,例如在Ⅱ类DMF中,除了原料药,还包括原料药的中间体以及生产过程中使用的物料或制剂。相比之下,我国的原料药备案仅限于化学原料药,生物制品方面仅涉及胰岛素类产品。由于我国“DMF”登记范围较为局限,导致国内一些本可通过备案成为DMF的GMP级别标准物料缺乏。由此,在进行生物活性关键物料控制时,我国研究者大量重复建设生产线和重复性研发注册工作。
未来我国可能也会借鉴美国的这一做法,以优化本国的生物制品药物申报流程,提高审评效率。
生物药物已经有相关研究者、关键物料供应商和CDMO平台公司走在前面,开始了GMP合规化之路,部分高质量基因编辑蛋白、抗体、细胞因子、关键试剂等领域的关键物料已经进行DMF文件化进程,一些企业的部分产品完成了美国FDA 的关键物料DMF备案,如江苏申基生物,已完成了N1-Me-Pseudo-UTP、CAP GAG & CAP GAG ( 3'OMe )和ATP& CTP & GTP多款mRNA原料产品的FDA 的DMF备案。
在生物药物领域全面正规化合规化的时代大潮下,合肥华纳生物医药科技有限公司做为一家具有核心技术的基因诊疗试剂、小核酸药物、合成生物学、荧光及化学发光染料等核心原料产品的科技创新产业平台,核心领导层提前进行全品种DMF注册的战略布局,制定了核心产品的GMP合规化战略方针,以点带面的形式开展公司质量管理体系的升级和强化,深化产品质量管理,以先进工艺结合先进管理的方式,制造出世界先进水平的核苷单体。
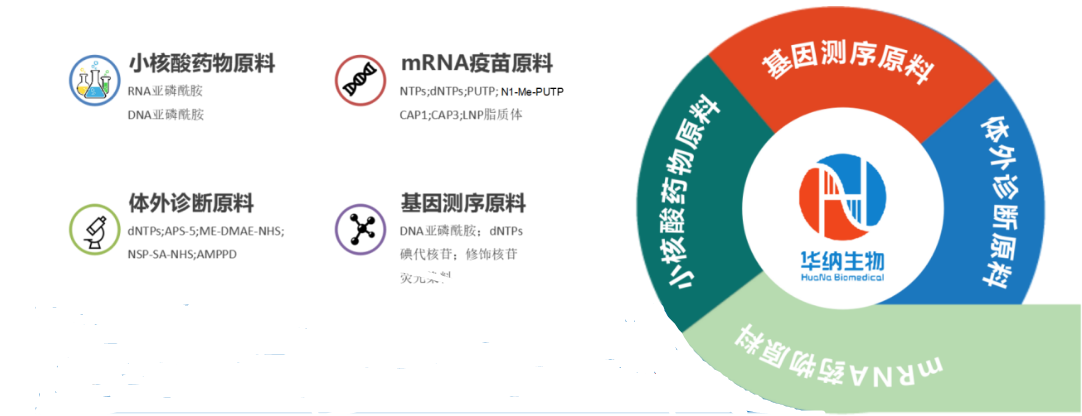
寡核苷酸和核苷单体是核酸原料药的关键物料,而符合质量要求的合格供应商在全球范围内都较为稀有。这主要源于工艺开发、工艺放大和质量控制上存在较高壁垒,且配套设备、洁净环境等前期投入非常大,同时生产需符合国际GMP要求。华纳生物的核苷单体中的N4-Ac-2'-O-Me-5'-O-DMT-C-3'-CE-Phosphoramidite,做为核苷单体中的代表产品,在市场上具有突出的品质优势,工艺形成成套的体系文件,杂质谱研究透彻,完成一套全面的杂质标准品,DMF文档也在逐步升级,可以满足优质客户的DMF申报资料需求,达到DMF的GMP级别标准物料,缩短了客户药物上市申报进程的时间,解决困扰客户的一大难题。
华纳生物做为上游化学平台,核酸药物原料不仅具有严格的来料检验标准,且经过严格理化生内控指标,做到人、机、料、法、环全方位质量管理体系把控,原料产品质量稳步提高!华纳生物将继续潜精研思,做好核酸药物原料正规化的先锋,努力打造更优质的产品与服务,促进核酸药物研发与商业化进程!
2022年10月31日,国家药品监督管理局食品药品审核查验中心颁布了《细胞治疗产品生产质量管理指南(试行)》。该指南特别强调了治疗性起始物料的GMP资质问题,明确指出直接用于细胞产品生产的基因修饰载体或其他功能性材料(例如病毒、质粒、RNA等)的生产、检验和放行流程必须符合GMP及其相关附录的严格标准。
然而,国内当前的研发和注册环境中,研究者在选择病毒、质粒、RNA等关键物料的GMP级别时面临诸多限制。由于许多研究者对GMP级别物料的重要性认识不足,他们在注册过程中可能会随意选择关键物料用于临床药品生产。
我国生物药物的研究者向CDE(国家药品监督管理局药品审评中心)提交生物制品药物申报,如比如临床研究申请(IND)、新药注册(NDA)和生物制品许可证申请(BLA)等,必须遵循CTD(通用技术文档)格式来整理注册文件。这些文件需要提供药物在安全性、有效性和质量三大方面的详尽信息,涵盖原液生产工艺、制剂生产工艺以及关键活性物料的相关技术细节。然而,准备这些材料不仅费时费力,而且由于生物制品原液生产工艺的复杂性,涉及众多上下游工艺中的关键物料,这些物料的GMP资质问题往往成为申报过程中的一大难题。
缺乏关键物料的GMP认证资质渠道会严重拖慢临床申报的进度,为应对这一挑战,美国FDA(食品药品监督管理局)建立了生物制品关键物料的DMF(药品主文件)备案体系。通过这一体系,生物制品的研究者可以将原液生产所需的关键物料技术信息,以DMF文件的形式直接提交给FDA进行备案并获得备案编号。研究者在生物药研究项目中需要提交FDA临床申请或上市注册的监管备案文件时可直接引用相关DMF编号,从而节约产品审查和评估时间,缩减新药临床申报的准备工作,这有力促进了研究者的药物上市申报进程。
从DMF备案登记的范围来看,美国的DMF适用范围相对较广,例如在Ⅱ类DMF中,除了原料药,还包括原料药的中间体以及生产过程中使用的物料或制剂。相比之下,我国的原料药备案仅限于化学原料药,生物制品方面仅涉及胰岛素类产品。由于我国“DMF”登记范围较为局限,导致国内一些本可通过备案成为DMF的GMP级别标准物料缺乏。由此,在进行生物活性关键物料控制时,我国研究者大量重复建设生产线和重复性研发注册工作。
未来我国可能也会借鉴美国的这一做法,以优化本国的生物制品药物申报流程,提高审评效率。
生物药物已经有相关研究者、关键物料供应商和CDMO平台公司走在前面,开始了GMP合规化之路,部分高质量基因编辑蛋白、抗体、细胞因子、关键试剂等领域的关键物料已经进行DMF文件化进程,一些企业的部分产品完成了美国FDA 的关键物料DMF备案,如江苏申基生物,已完成了N1-Me-Pseudo-UTP、CAP GAG & CAP GAG ( 3'OMe )和ATP& CTP & GTP多款mRNA原料产品的FDA 的DMF备案。
在生物药物领域全面正规化合规化的时代大潮下,合肥华纳生物医药科技有限公司做为一家具有核心技术的基因诊疗试剂、小核酸药物、合成生物学、荧光及化学发光染料等核心原料产品的科技创新产业平台,核心领导层提前进行全品种DMF注册的战略布局,制定了核心产品的GMP合规化战略方针,以点带面的形式开展公司质量管理体系的升级和强化,深化产品质量管理,以先进工艺结合先进管理的方式,制造出世界先进水平的核苷单体。
寡核苷酸和核苷单体是核酸原料药的关键物料,而符合质量要求的合格供应商在全球范围内都较为稀有。这主要源于工艺开发、工艺放大和质量控制上存在较高壁垒,且配套设备、洁净环境等前期投入非常大,同时生产需符合国际GMP要求。华纳生物的核苷单体中的N4-Ac-2'-O-Me-5'-O-DMT-C-3'-CE-Phosphoramidite,做为核苷单体中的代表产品,在市场上具有突出的品质优势,工艺形成成套的体系文件,杂质谱研究透彻,完成一套全面的杂质标准品,DMF文档也在逐步升级,可以满足优质客户的DMF申报资料需求,达到DMF的GMP级别标准物料,缩短了客户药物上市申报进程的时间,解决困扰客户的一大难题。
华纳生物做为上游化学平台,核酸药物原料不仅具有严格的来料检验标准,且经过严格理化生内控指标,做到人、机、料、法、环全方位质量管理体系把控,原料产品质量稳步提高!华纳生物将继续潜精研思,做好核酸药物原料正规化的先锋,努力打造更优质的产品与服务,促进核酸药物研发与商业化进程!



